
Robbins, Liu & Kelly present RECUR, a method for identifying recurrent amino acid substitutions from multiple sequence alignments that is fast, easy to use, and scalable to thousands of sequences.
#compbio
Deng et al. present TreeProfiler, a tool for automated annotation and interactive exploration of hundreds of features along large gene and species trees, with seamless summarization of mapped traits at internal nodes.
Martí-Gómez et al. developed gpmap-tools, integrating models for inference, phenotypic imputation, and error estimation from multiplex assays of variant effect data or natural sequences in the presence of genetic interactions.
Anchieri et al. benchmark the inference of selection with aDNA-like time series datasets, showing that ApproxWF can accurately estimate selection with datasets of ∼100 individuals when selection is strong.
Ramos-González et al. present PharaohFUN, a web application designed for the evolutionary and functional analysis of protein sequences in photosynthetic eukaryotes, leveraging orthology relationships.

Malik et al. present the web-based Structome-AlignViewer, for evaluating structure-aware alignments through spatial mapping of alignment columns to protein structures, and quantitative confidence scoring.
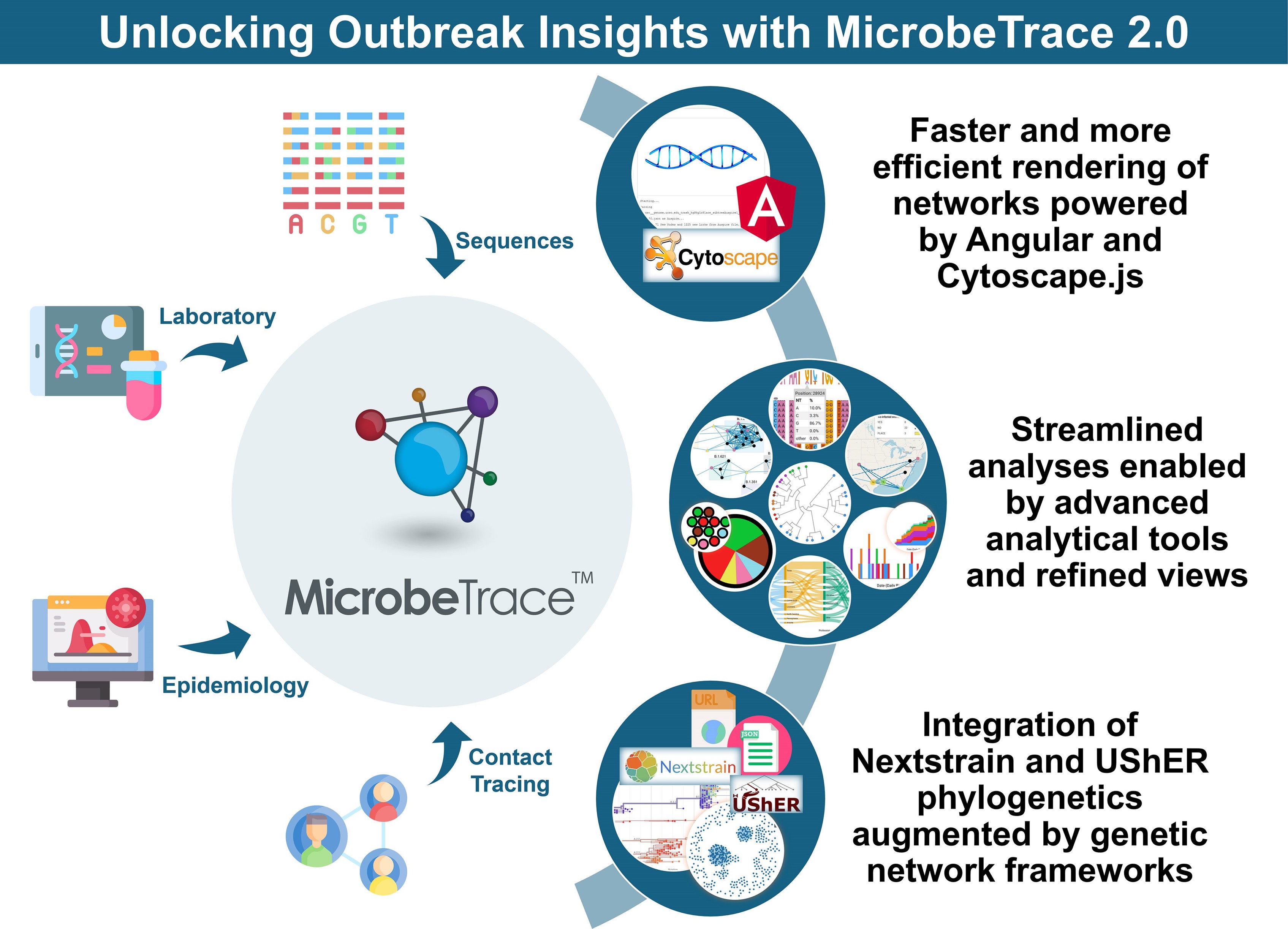
Shankar et al. present the updated MicrobeTrace 2.0 as a next-generation, interoperable tool for genomic epidemiology and data-driven public health response.

In 1952, Alan Turing proposed that simple chemical reactions + diffusion could create spots, stripes, and spirals in nature. 70 years later, we're still finding his equations everywhere—from zebrafish stripes to cardiac arrhythmias. Math predicting biology at its finest.
In a new Review, Peede et al. overview the SMC model and extensions, discuss examples of discoveries made with the help of SMC-based inference, and comment on the assumptions, benefits, and drawbacks of various methods.

Thawornwattana, Rannala & Yang use simulation to study the false positive rate of a Bayesian test of gene flow under the MSC with multiple influencing factors.
Haller, Ralph & Messer present SLiM 5, a major extension of the SLiM simulation framework for simulating multiple chromosomes, enabling a heightened level of realism for full-genome simulations.

#compchem #compbio Good read: Fast Parametrization of Martini3 Models for Fragments and Small Molecules https://pubs.acs.org/doi/10.1021/acs.jctc.5c01178

#compchem #compbio Last preprint of the year: "Fast, systematic and robust relative binding free energies for simple and complex transformations : dual-LAO".
https://arxiv.org/abs/2512.17624
Great work by N. Ansari.
(qubit-pharma). Another nice collab with J. Hénin.

💫 We just released the weights of the #FeNNixBio1 foundation machine learning model for drug design! 💫
Weights: https://github.com/FeNNol-tools/FeNNol-PMC
FeNNol code: https://github.com/FeNNol-tools/FeNNol
The models are distributed under the open source ASL license (non-commercial academic research). #compchem #compbio
To address uncertainty in model choice for reconstructing population history, Xu et al. introduce a new Bayesian model averaging (BMA) framework.
Mirchandani et al. introduce Callable Loci And More (clam), a tool that leverages callable loci to accurately estimate population genetic statistics (π, dxy, and FST).
Szymanski et al. developed quicksand, an open-source Nextflow pipeline designed for rapid and accurate taxonomic classification of mammalian mitochondrial DNA in sedimentary ancient DNA samples.

Can't wait to read this thriller by Carsten Kutzner that dropped: "Scaling of the GROMACS Molecular Dynamics Code to 65k CPU Cores on an HPC Cluster" /hj
In seriousness, this sounds like a really cool, and difficult, problem! Looking forward to seeing how they approached it.
#fediscience #compbio
Huang et al. present SAI, a Python package for computing introgression statistics, and demonstrate its application in two datasets.
Rahman, Smith & Szpiech present selscan v2.1, a dynamic programming algorithm to improve runtime and memory usage in the calculation of extended haplotype homozygosity.
Client Info
Server: https://mastodon.social
Version: 2025.07
Repository: https://github.com/cyevgeniy/lmst