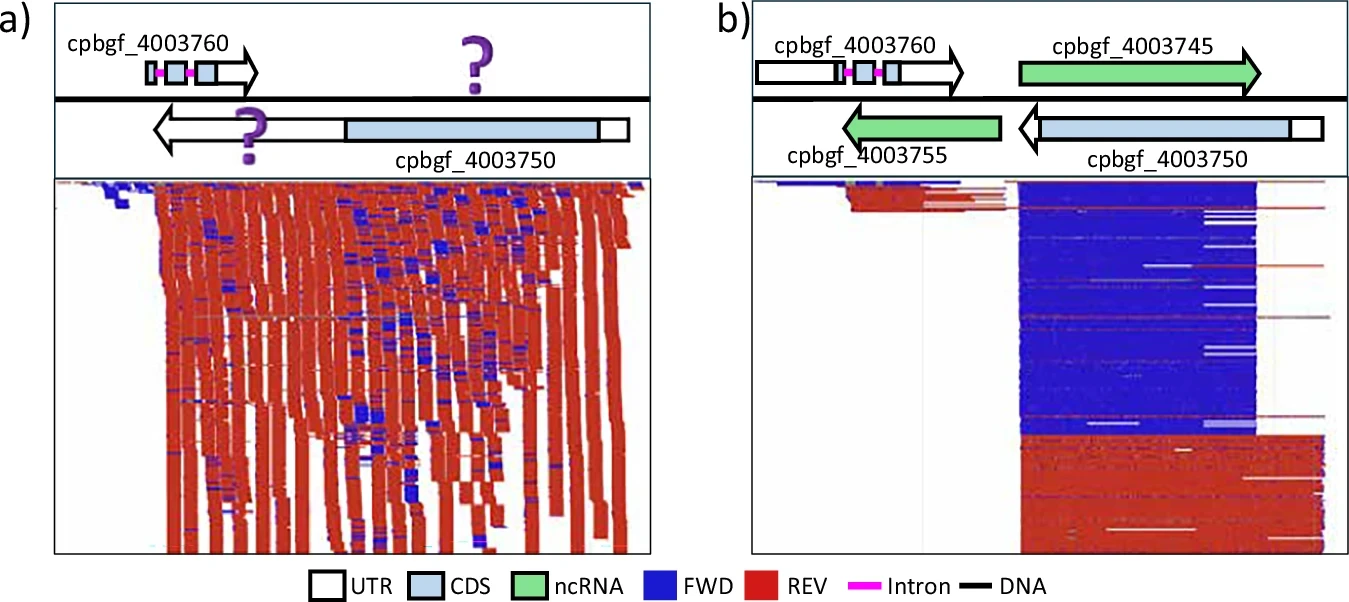
My PhD student authored a rather niche piece of software written in Rust that introduces a new type of index for BAM files, allowing fast counting and filtering of reads based on their FLAG attributes:
Although we mention possible usage scenarios, we are far from sure how frequent, or useful these might be. Would be grateful for any input on this from the community, especially from people who can imagine including #bafiq in their pipelines or BAM file repositories. Negative examples might also help.
#bioinformatics #genomics #sequencing #genome #samtools